I got your fertile soils right here...
There
are a lot of ways that an environment can be extreme,
but today, we’ll focus on chemical extremes: pH (acid and alkaline),
and salinity. Believe me, this will keep us busy for quite a while.
Let’s start with pH. Extreme pH can be defined as anything falling outside the range of 5.5 to 6.5, which is where the bulk of plants do best. Plenty of commonplace graden plants like azaleas, dogwoods and blueberries thrive at a more acid pH, often in the 5.0 - 5.5 range. Once soil pH dips below 4.5, the soil is considered “strongly acid”, and most plants begin to suffer from the acidity (for further detail, wander back to this post), mostly in the form of nutrient deficiencies. Simplifying matters greatly, let’s leave it at “nutrient uptake is impaired at low pH”.
Let’s start with pH. Extreme pH can be defined as anything falling outside the range of 5.5 to 6.5, which is where the bulk of plants do best. Plenty of commonplace graden plants like azaleas, dogwoods and blueberries thrive at a more acid pH, often in the 5.0 - 5.5 range. Once soil pH dips below 4.5, the soil is considered “strongly acid”, and most plants begin to suffer from the acidity (for further detail, wander back to this post), mostly in the form of nutrient deficiencies. Simplifying matters greatly, let’s leave it at “nutrient uptake is impaired at low pH”.

And how!
Most plants, but not all. Some plants do very well at extremely low pH - river birch (Betula nigra) is known to enjoy pHs as low as 2, while pitch pine (Pinus rigida) in addition to tolerating a fire regime tolerates soils down to a pH of 3.4. How do they do it?
Well, remember from the last time we took a look at acid soils, there are two main problems to deal with. One, nutrient availability is low. Two, aluminum (Al) availability is high (and aluminum is toxic). These problems have a common solution - mycorrhizae!
Well, remember from the last time we took a look at acid soils, there are two main problems to deal with. One, nutrient availability is low. Two, aluminum (Al) availability is high (and aluminum is toxic). These problems have a common solution - mycorrhizae!
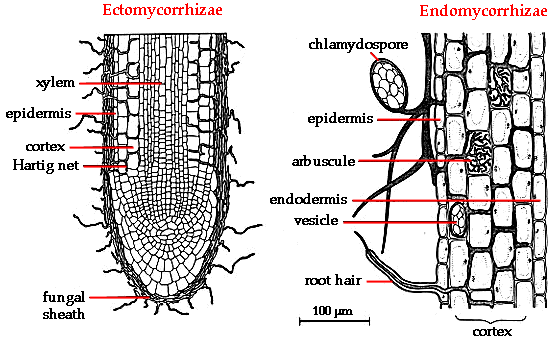
From left to right, the art view and the technical view
Mycorrhizae
are a family of fungi that grow in a symbiotic association with many
plant species. Mycorrhizae do all kinds of cool things, including making
nutrients more accessible. The explanation is simple - associating with
a mycorrhizal network gives plants access to a huge amount of soil,
without having to put out all of those roots. When nutrient uptake is
limited or nutrient concentrations are low, having access to more soils means more nutrient uptake.
Ok, so that should solve the low nutrient problem, but shouldn’t it
also increase the Al problem? Not quite. Acid tolerant plants have been shown
to block out Al with mycorrhizae - rather than be brought into the
plant, Al accumulates harmlessly on the surface of the plant-mycorrhizae
juncture.
So, one end of the pH spectrum can be managed, with a little help from some fungal friends. What about the other end of the spectrum, the high-pH, alkaline soils? As that image of nutrient availability shows, limitations kick in at higher pH as well - iron (Fe), for example, is often less accessible. So, some successful alkaline-tolerant plants have evolved efficient nutrient usage to limit any limitations. Mycorrhizae aren’t just for acid soils, either - plants growing in alkaline soils have been found to benefit from the increased nutrient access made possible by such a symbiotic fungal relationship.
So, one end of the pH spectrum can be managed, with a little help from some fungal friends. What about the other end of the spectrum, the high-pH, alkaline soils? As that image of nutrient availability shows, limitations kick in at higher pH as well - iron (Fe), for example, is often less accessible. So, some successful alkaline-tolerant plants have evolved efficient nutrient usage to limit any limitations. Mycorrhizae aren’t just for acid soils, either - plants growing in alkaline soils have been found to benefit from the increased nutrient access made possible by such a symbiotic fungal relationship.
Just
like acid soils are plagued by both low nutrient availability and high
Al concentrations, so alkaline soils have more than limited access to Fe
to contend with. Here, the problem isn’t Al, but rather, salt.
Extremely alkaline soils (pH of above 9) are the result of accumulated sodium carbonate
- while salt is not the main limiting factor in such soils, plants must
content with the salt to thrive. Remember, as we saw in our examination
of dune plants, salt is not a friend to all living things.
Plants in alkaline soils have been shown to excrete salts accidentally
taken up, or to store water within the leaf in such a way as to dilute
salts.
This makes a great segue to saline soils. Saline soils are (duh) saline, but not necessarily due to high concentrations of sodium. High concentrations of calcium, magnesium, potassium, chloride, and bicarbonate, among other things, can make a soil saline. Saline soils can be directly toxic to plants - chloride and sodium are both capable of damaging plant cells and tissues. Nutrient deficiencies are also an issue - high concentrations of the saline ions can make it more difficult to bring up actual nutrients, and sodium actively impairs nutrient uptake. Then there is the water issue.
This makes a great segue to saline soils. Saline soils are (duh) saline, but not necessarily due to high concentrations of sodium. High concentrations of calcium, magnesium, potassium, chloride, and bicarbonate, among other things, can make a soil saline. Saline soils can be directly toxic to plants - chloride and sodium are both capable of damaging plant cells and tissues. Nutrient deficiencies are also an issue - high concentrations of the saline ions can make it more difficult to bring up actual nutrients, and sodium actively impairs nutrient uptake. Then there is the water issue.
What a wonderful place to be a plant.
There are a number of controls on how water moves in an environment, one of these controls being the concentration gradient. In brief, water moves down a gradient of solute concentration - water with lower concentrations of solutes moves towards regions of higher solute concentration, and eventually reaches a uniform solute concentration. This becomes a problem when in order to survive, a plant needs to maintain a lower solute concentration in water within the root - water within the root is attracted to the highly alkaline water outside of the root. Additionally, when the plant goes to take up water, it is forcing water to move in the opposite direction of the gradient. So, how do salt-tolerant plants cope with this?
Actually, there a couple ways to do this, including a really complicated arrangement of cell membranes in the roots, but to my mind, the most elegant solution is to create an osmotic gradient within the plant. Certain plants sequester either organic compounds or ions from the saline soils within certain parts of the plant, at greater concentrations than the ions within the soils. This has the effect of making the concentration of solutes higher within the plant than without, and driving water into the plant. It takes less energy for the plant to use ions taken up from the soil solution, and this also has the effect of harmlessly sequestering salts brought into the plant. Pretty cool, huh?
This is only scratching the surface of the extreme environments plants laugh at. In future posts, we'll look at plants which thrive in freezing temperatures, drought conditions, and unbelievably windy mountaintops. Not to mention the bacteria out there that regard acid mine drainage, petroleum derivatives and toxic chemical spills as a delicious breakfast buffet. Excited yet?